
Evaluating the Boltz-2 Co-folding Model for Challenging Modalities: A Two-Case Study with Covalent Inhibitor and Macrocyclic Molecular Glue
Published:
Introduction
In the rapidly evolving landscape of drug discovery, Artificial Intelligence (AI) co-folding models have gained significant attention, frequently highlighted in public reports for their potential to revolutionise structure-based drug design (SBDD). These computational tools promise to accelerate the identification and optimisation of novel drug candidates by predicting the intricate interactions and corresponding conformations between receptors and ligands, under induced fit effects. However, despite the widespread enthusiasm, there remains a notable gap in detailed performance investigations, particularly concerning challenging modalities such as covalent inhibitors and the emerging class of molecular glues. For the pharmaceutical industry, where real-world applications demand robust and reliable predictive capabilities, evaluating these models against those complex cases is crucial. This helps us to understand not only what co-folding models can achieve but also their current limitations that need to be improved.
In this LinkedIn blog, I would like to share my first-time experience using Boltz-2 to predict the structures of two special protein-ligand complexes: a covalent kinase inhibitor and a macrocyclic molecular glue.
Key Conclusions at the Outset
The co-folding model demonstrated satisfactory performance on a well-known protein target with a covalent ligand from its training set, accurately reproducing the experimental structure based on its confidence scores.
Generative prediction appears sensitive to different 3D inputs of the chemical component dictionary (CCD) for the same ligand. More reasonable results often come from the default or bioactive embedding.
For novel chemical modalities, AI models struggle to generalise and predict corresponding structures within protein complexes. The conversion of stereochemistry is a critical issue, often manifesting as “hallucinations” in the diffusion model.
Some improvements were observed when incorporating Gaussian denoising with physical constraints or steering potentials. This approach could yield more reasonable structures with correct ligand stereochemistry, protein complementarity, and low-energy conformations.
The diversity of conformational sampling remains a bottleneck for data-driven deep learning architectures. This limitation could potentially be addressed by complementing the models with physical simulations based on the energy.
Co-folding for a Covalent Kinase Inhibitor Complex
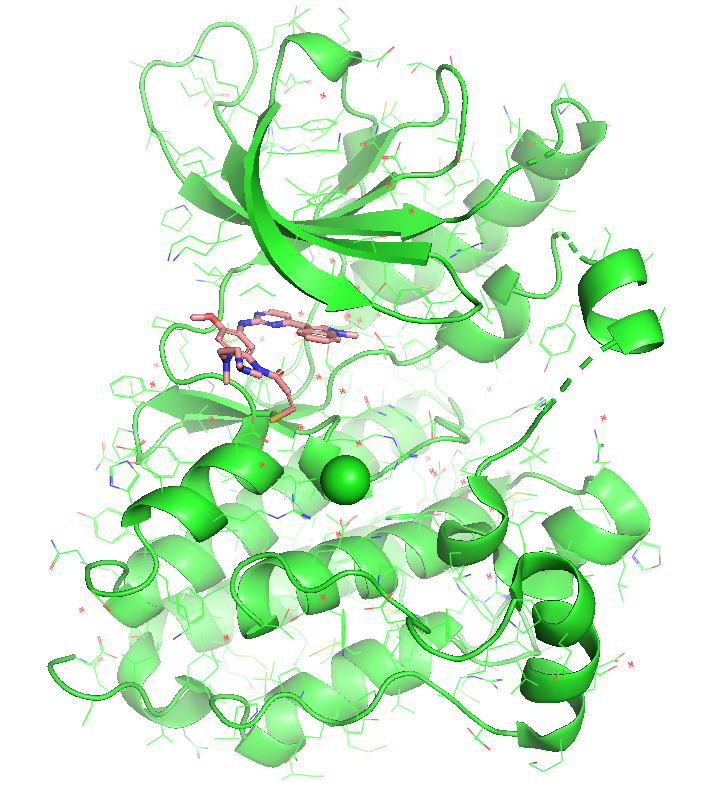
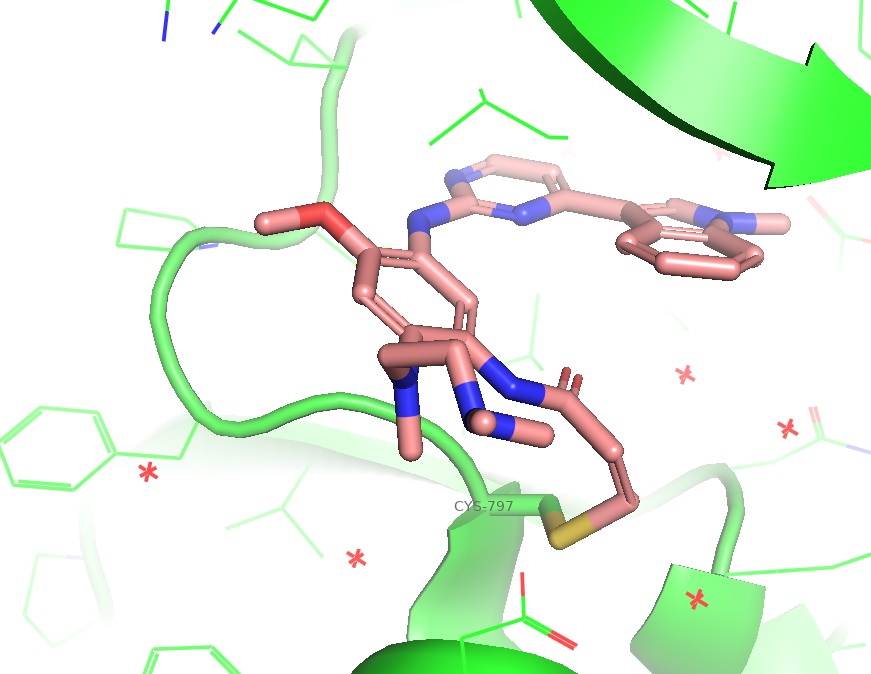
Figure 1. The co-crystal structure of EGFR kinase domain (WT) in complex with Osimertinib (PDB: 6JXT). The ligand acrylamide warhead form a Michael adduct with CYS-797 for an irreversible covalency.
sequences:
- protein:
id: [A]
sequence: LTPSGEAPNQALLRILKETEFKKIKVLGSGAFGTVYKGLWIPEGEKVKIPVAIKELREATSPKANKEILDEAYVMASVDNPHVCRLLGICLTSTVQLITQLMPFGCLLDYVREHKDNIGSQYLLNWCVQIAKGMNYLEDRRLVHRDLAARNVLVKTPQHVKITDFGLAKLLGAEEKEYHAEGGKVPIKWMALESILHRIYTHQSDVWSYGVTVWELMTFGSKPYDGIPASEISSILEKGERLPQPPICTIDVYMIMVKCWMIDADSRPKFRELIIEFSKMARDPQRYLVIQGDERMHLPSPTDSNFYRALMDEEDMDDVVDADEYLIPQQGFFSSPSTSRTPLLSSLSATSNNSTVACIDRNGLQSCPIKEDSFLQRYS
- ligand:
id: [B]
ccd: YY3
constraints:
- bond:
atom1: [A, 106, SG]
atom2: [B, 1, C9]
Text 2. The input .yaml file as example. The covalency was defined through a bond constraint.
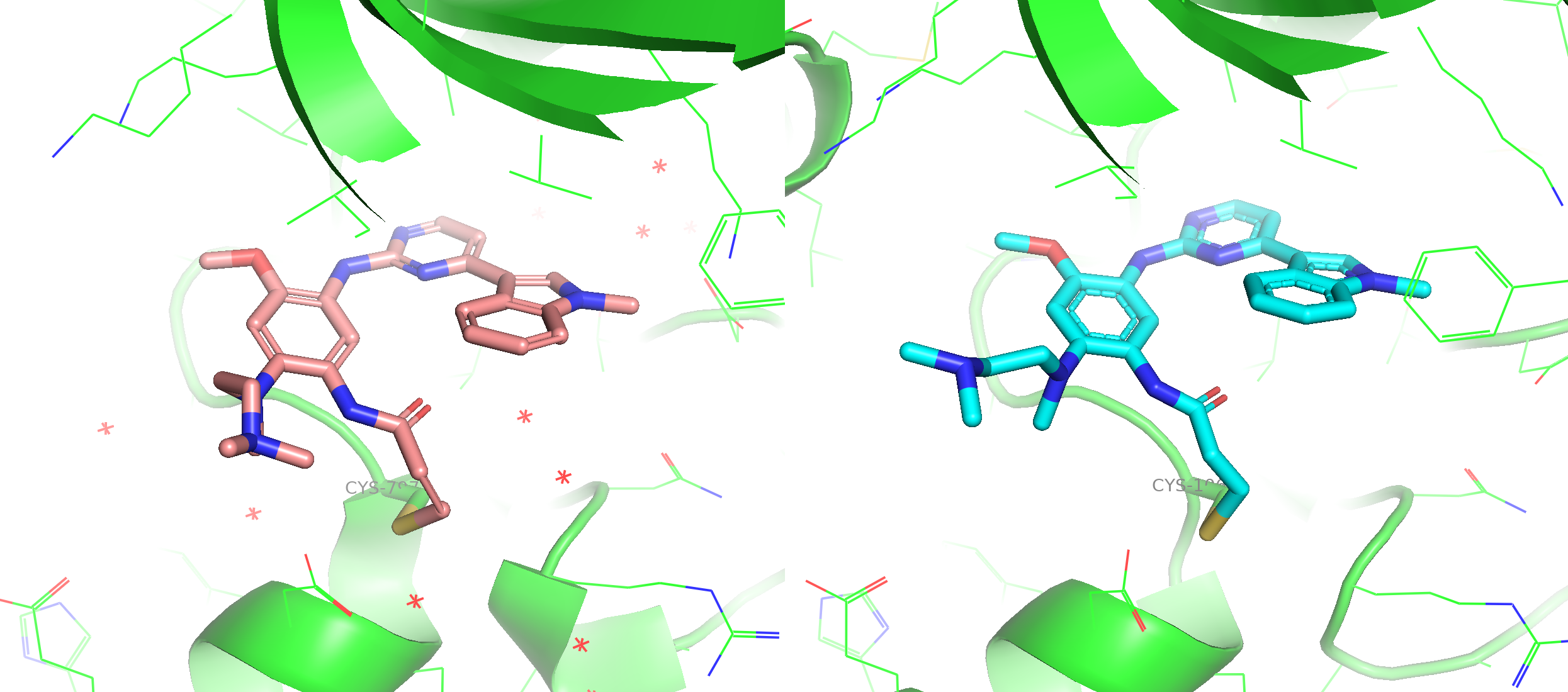
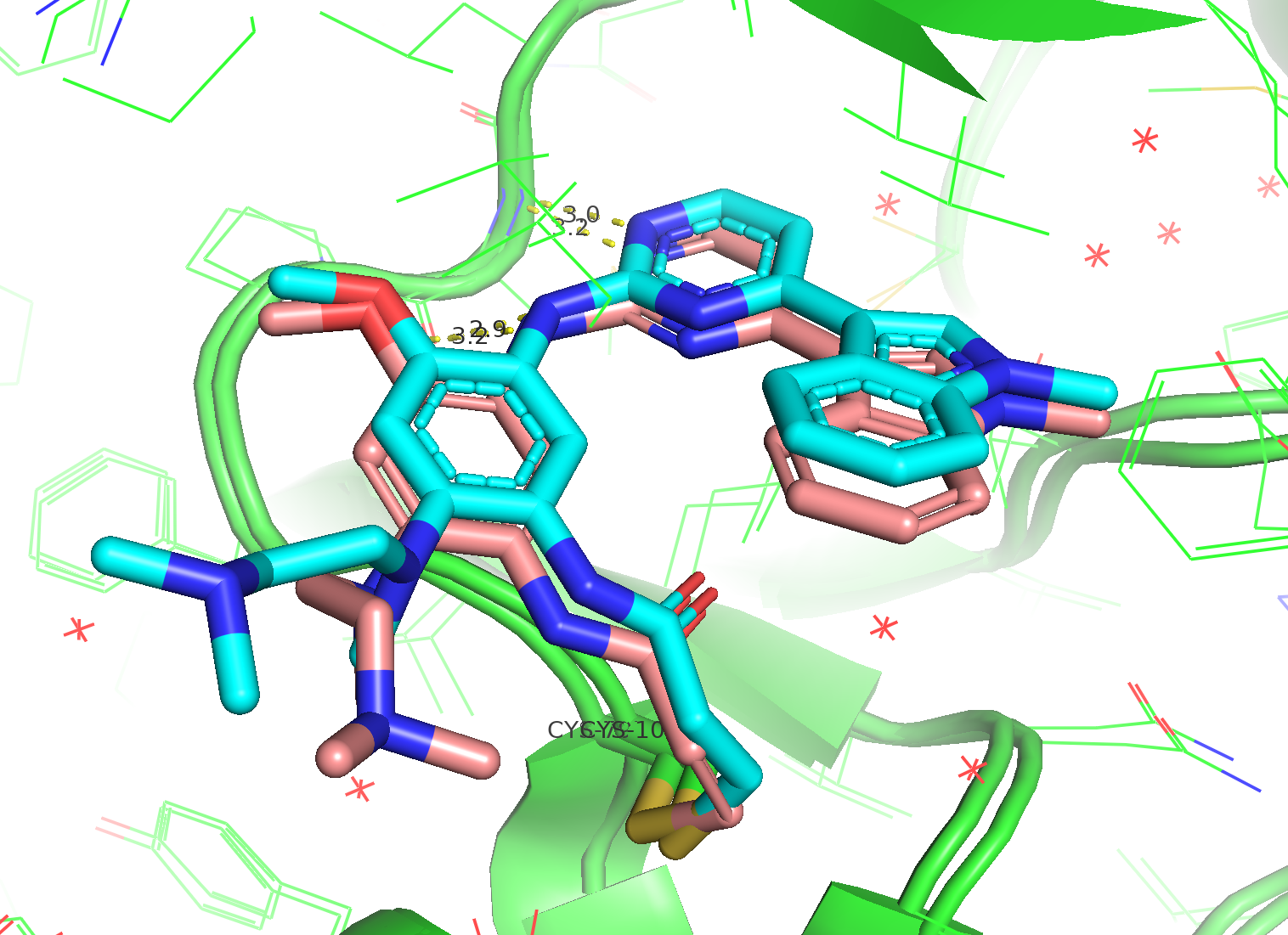
Figure 3. The alignment between co-crystal structure (6JXT, ligand in pink stick) and a predicted model from Boltz-2 (ligand in blue stick). The critical interaction with hinge loop was preserved by co-folding.
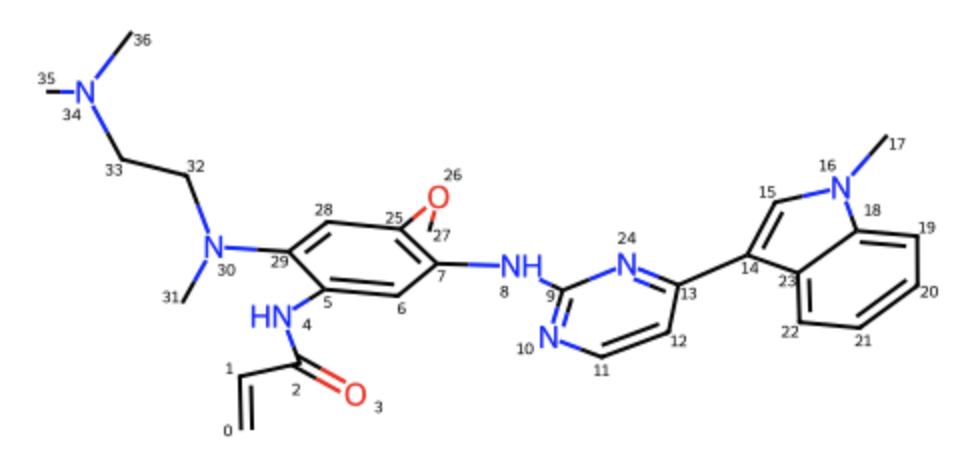
Figure 4. The RDKit mol object for Osimertinib with a random 3D conformer embedded (ETKDG with MMFF minimisation), which was used for the subsequent EGFR co-folding tests under covalent bond constraint. (now atom name index is C0)
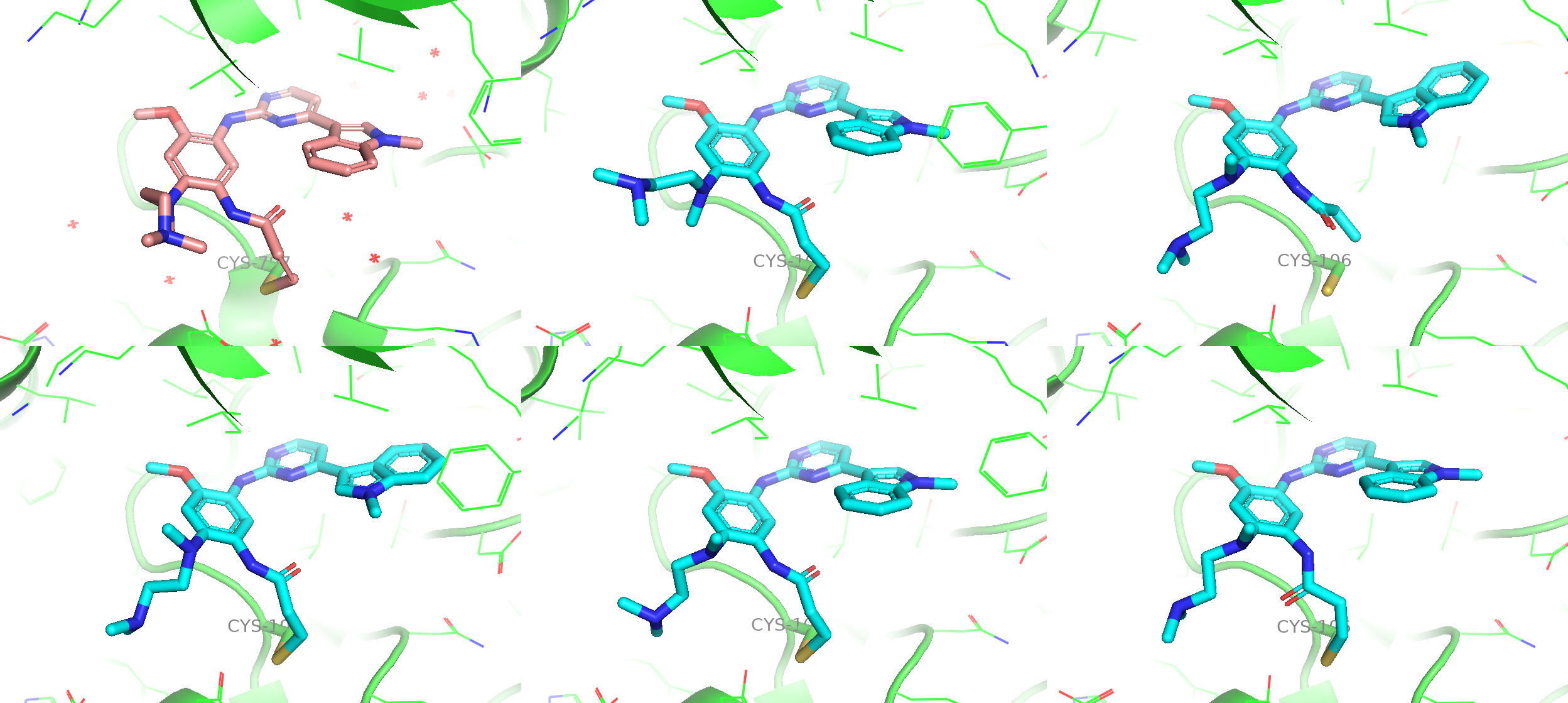
Figure 5a. The comparison among the co-crystal structure (upper left), a predicted model from default CCD (upper middle) and 4 more independent samples from customised CCD with high confidence scores.
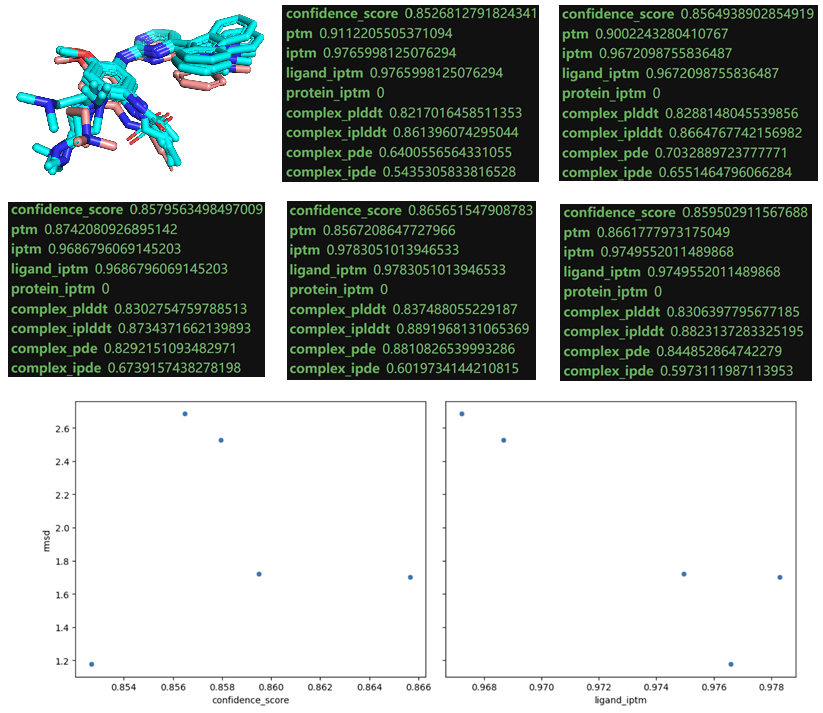
Figure 5b. The scoring matrices for 5 predicted models. A very weak signal was given by ligand iptm which indicates the reproducibility of experimental structure in this case.
Co-folding for a Macrocyclic Molecular Glue Complex
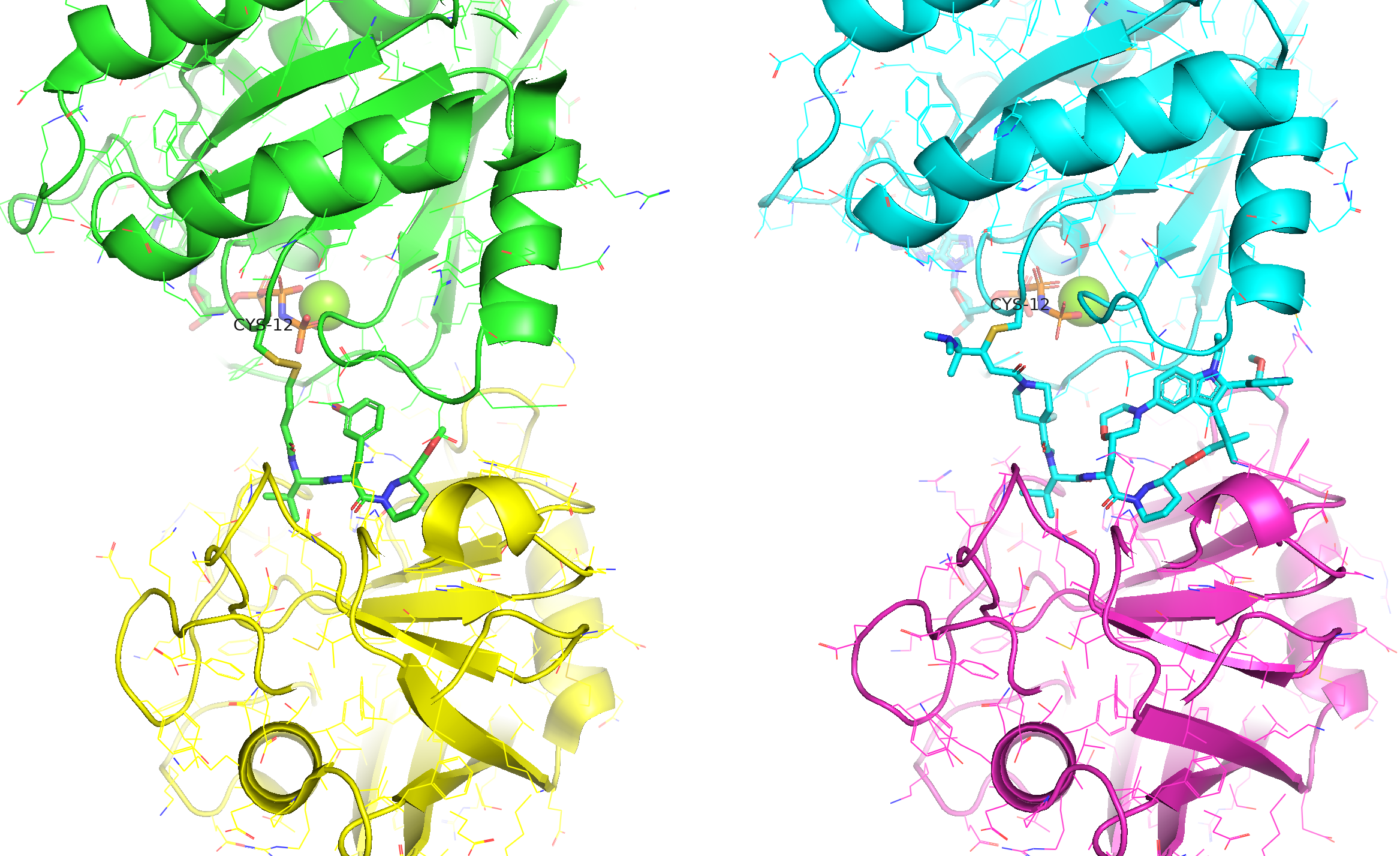
Figure 6. The covalent molecular glue for KRAS G12C (upper domain) and CYPA (lower domain). Two co-crystal structures were available respectively for a lead ligand (left, 8G9Q) and the optimised macrocycle - Elironrasib (right, 9BFX).
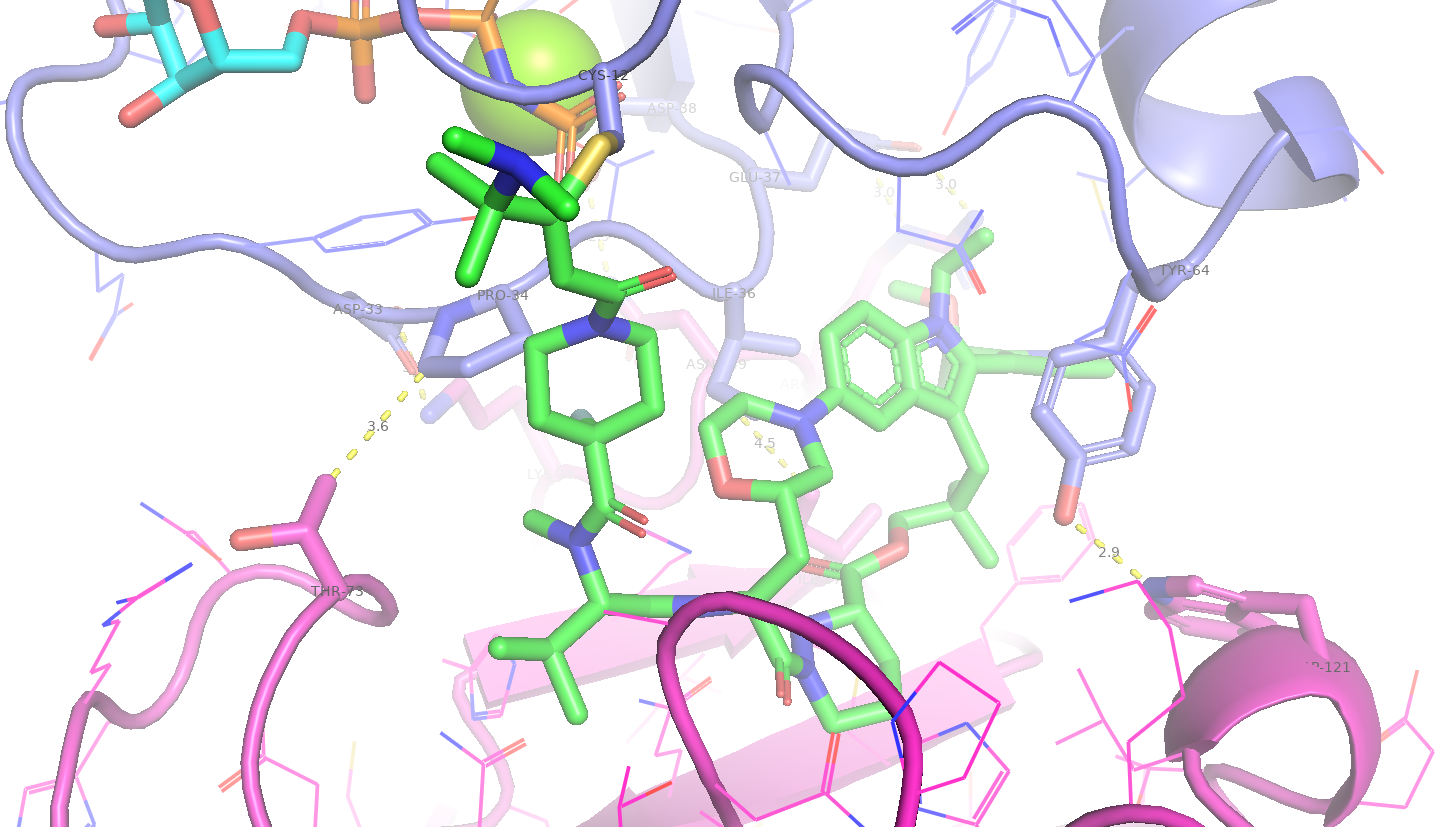
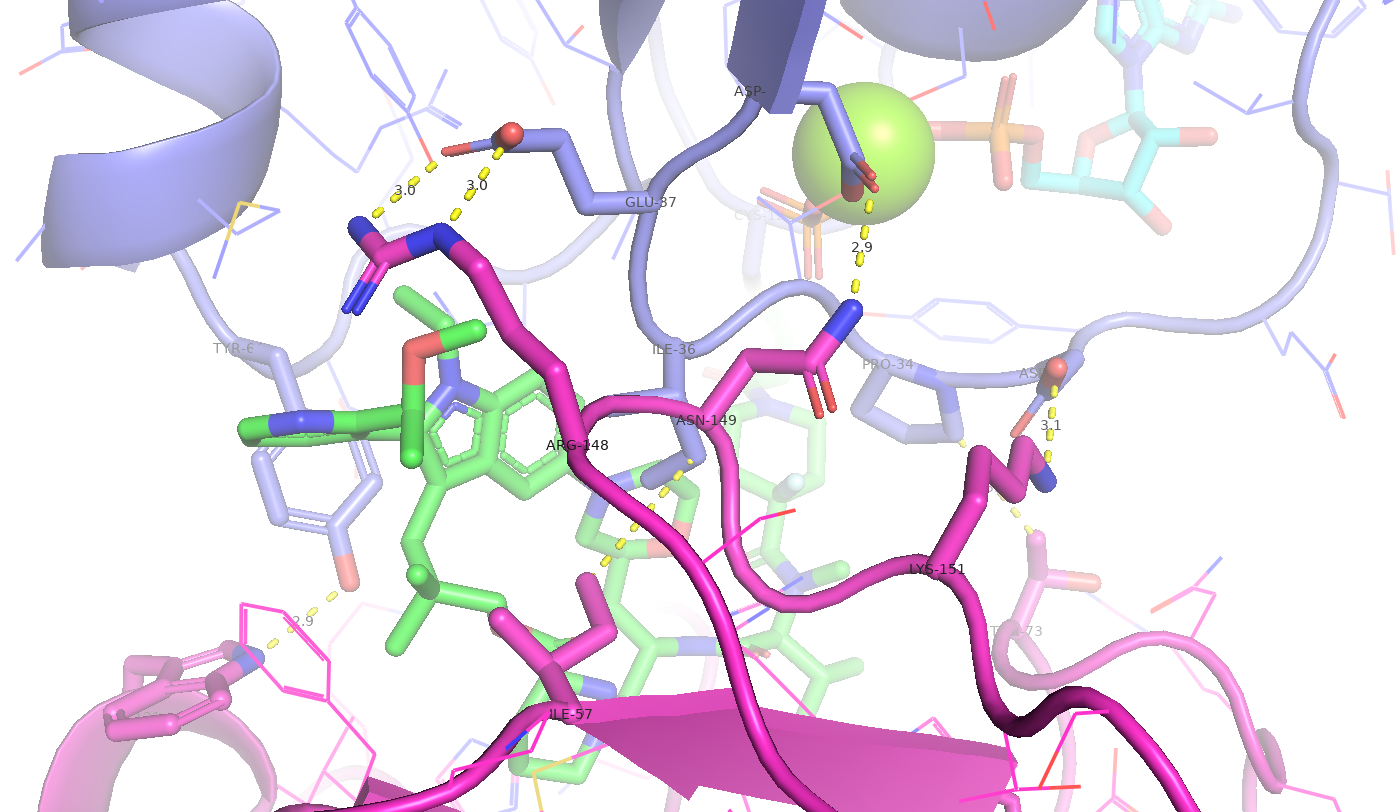
Figure 7. The Elironrasib-induced proximity between KRAS G12C and CYPA. Several hydrophobic interactions and electrostatic complementarities (H-bonds and salt bridges) were identified from the co-crystal structure.
For the subsequent co-folding prediction, I expected the Boltz-2 model to reproduce such ternary complex, especially for the proteins' orientation under enough sampling, since its training set contains 8G9Q. However, I was more doubtful about the AI's performance on organic macrocycles, given that chemical space is far more diverse than 20 amino acids and those cyclic peptides.
Traditionally, computational chemists would use Monte Carlo or Genetic Algorithm-based docking (rigid and ensemble) to tackle such problems, especially when there is any available or even confident holo structure (i.e., the relative orientation of the two proteins in the functional ternary complex.) for generating the grid box of receptor. Furthermore, advanced simulation techniques (e.g., enhanced MD sampling) with properly QM/DFT-parameterised force fields have been shown to reproduce NMR solution ensembles for macrocyclic drugs. These ensembles often include bioactive-like poses because the ring can constrain the dynamics of molecule into a narrow energy landscape, even without any receptor embraced. With above background, I was keen to see how an AI diffusion model would generate structures under circumstances where not much physics has been learned in this field.
Ligand Preparation and Protein Performance
To introduce diversity, I customised three different CCD objects for Elironrasib, ensuring each entry has correctly defined stereochemistry (Figure 8a). Diverse strategies were used to create the 3D input pickle files for same ligand (Figure 8b):
- A random conformer
- Few low-energy conformers (from 1000 steps of ETKDG search followed by MMFF minimisations)
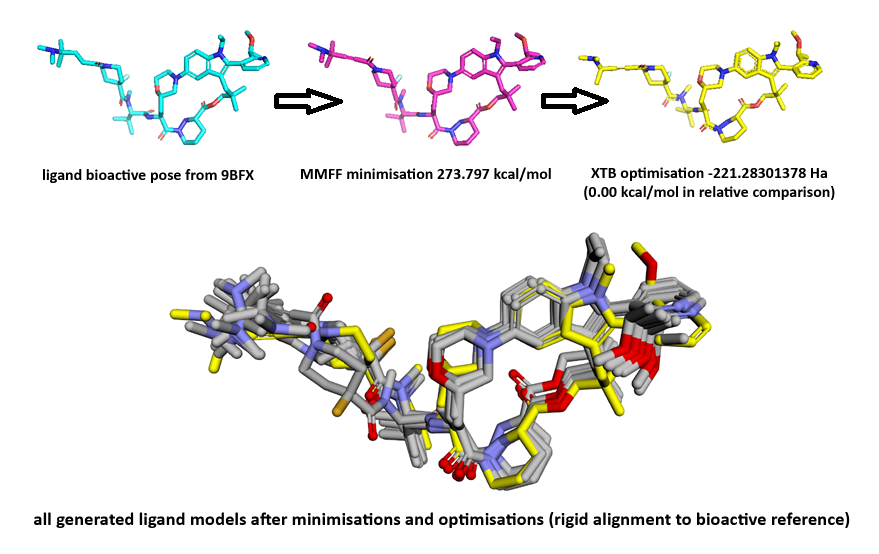
- The bioactive conformer (extracted from 9BFX and minimised to recover warhead geometry)
Notably the bioactive conformer with local minimisation was calculated to be a low-energy state by classic Merck molecular force field (MMFF), even more stable than those minima sampled from ETKDG algorithm.
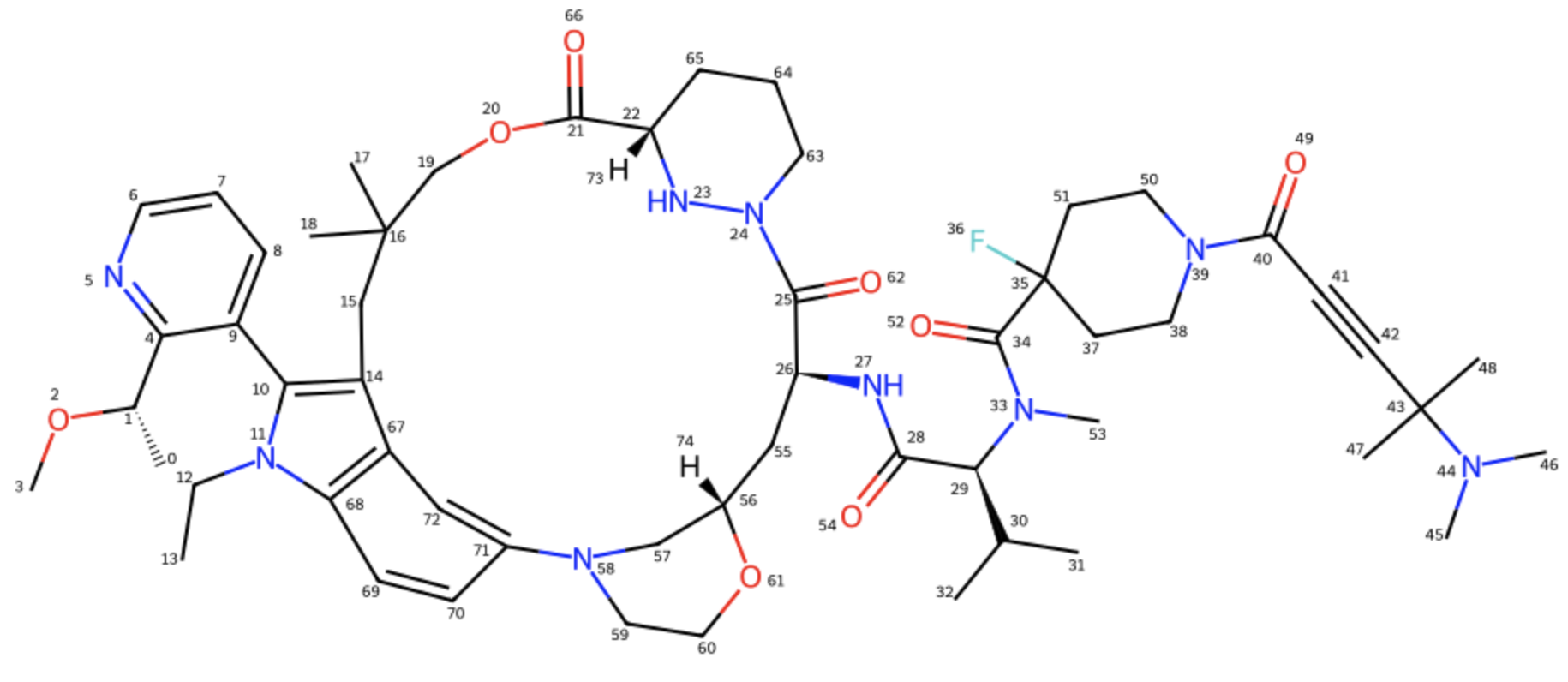
Figure 8a. The Elironrasib ligand in 2D with all correct stereochemistry labels clearly.
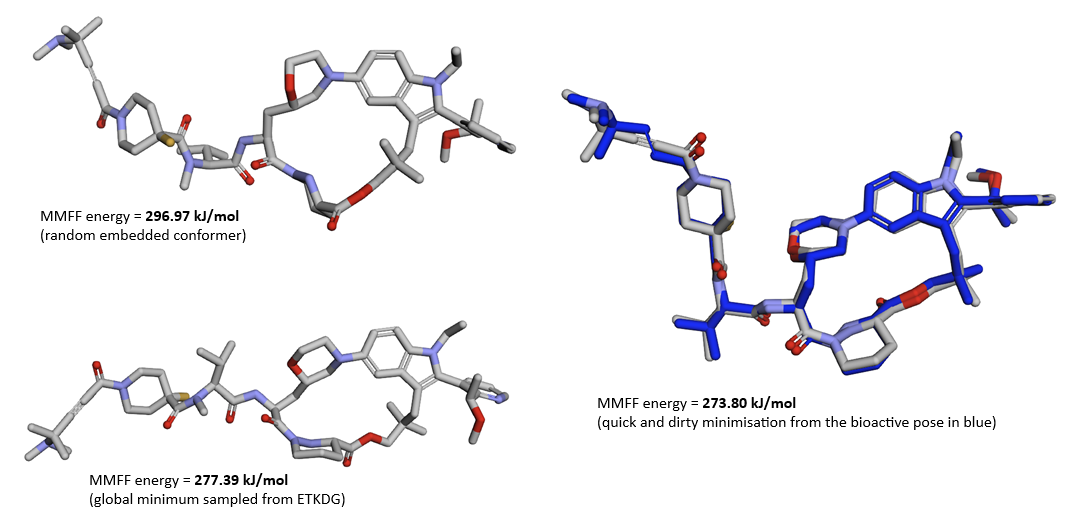
Figure 8b. Three different CCD entries of Elironrasib were used for the co-folding predictions of ternary complex, each possess a unique 3D conformer based on all correct stereochemistry. (Error note: all energy units should be kcal/mol in this figure)
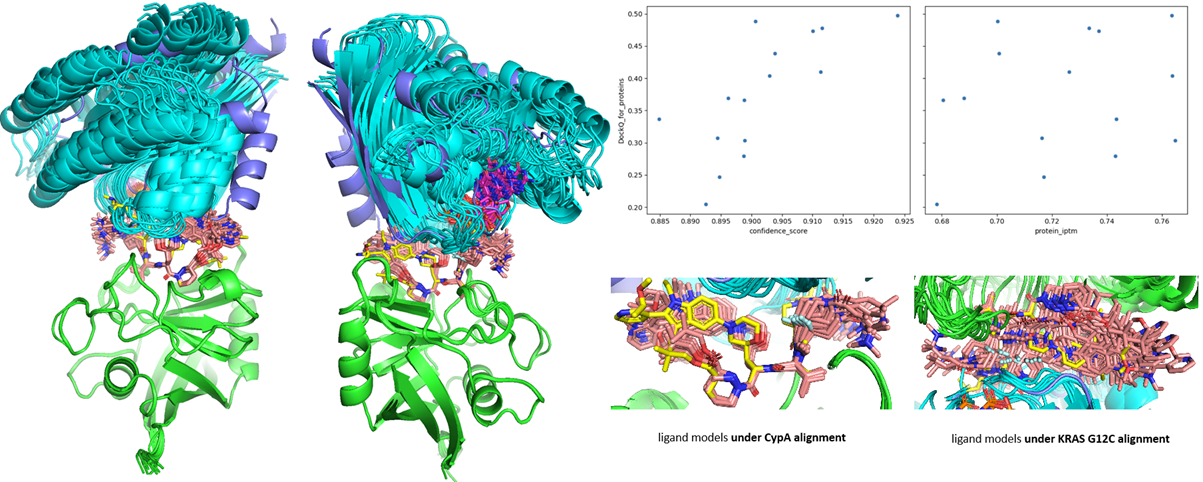
Figure 9. The ensemble of Boltz-2 co-folding prediction compared to 9BFX (ligand in yellow stick; KRAS in dark blue cartoon). All models are aligned by CYPA backbone (green cartoon) for clear demonstration. The models’ confidence and protein iptm scores are plotted with DockQ evaluation.
The Performance on Novel Macrocyclic Ligand
Imperfect Generalisation and Problematic Stereochemistry
Unfortunately, the model's performance on the chemical ligand itself was not as good as on protein receptors. The co-folding struggled to predict the most crucial regions where it was expected to generalize in this novel chemical space according to physical rules. If all ternary models are aligned by CYPA (mechanistically the first protein recruited and bound by Elironrasib as glue), the ligand geometries deteriorate as they approach the KRAS surface (Figure 9). This is even more apparent when aligning by KRAS, the subsequent target under induced covalency.
Out of 15 diffused samples, up to 8 ligand poses had the wrong chiral centres on the substituted morpholine ring (Figure 10a). This is the novel bridging moiety close to KRAS that was not present in the 8G9Q structure from the training set. For the remaining 7 poses with all correct stereocentres, none of their bi-aryl torsions were sampled properly. Again, this moiety was not in 8G9Q.
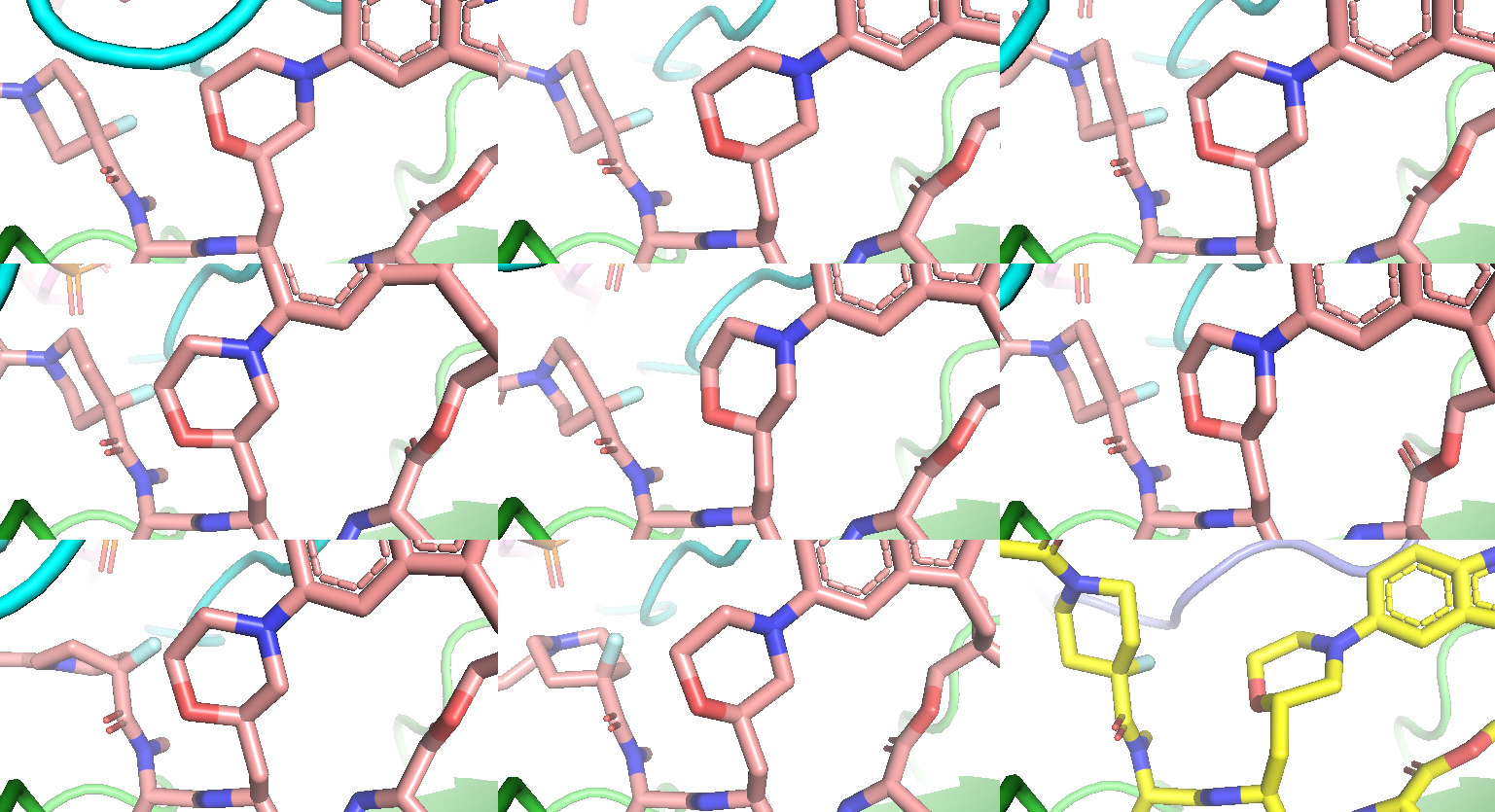
Figure 10a. 8 of 15 co-folding samples showing wrong stereochemistry on the morpholine substitution (pink stick), compared to 9BFX reference (yellow stick).

Figure 10b. Remaining 7 co-folding samples with correct stereochemistry. All models’ bi-aryl torsional profile is different from the experimental structure.
| Ternary sample index | All correct ligand stereochemistry | Ligand rmsd to the bioactive (CYPA alignment) | MMFF min. E relative to the bioactive (best rms) | XTB opt. E relative to the bioactive (best rms) |
|---|---|---|---|---|
| 3 | True | 3.77 Å | +3.95 kcal/mol (2.29 Å) | +2.12 kcal/mol (2.18 Å) |
| 6 | True | 3.90 Å | +1.94 kcal/mol (2.26 Å) | +1.28 kcal/mol (2.09 Å) |
| 7 | True | 4.09 Å | +8.08 kcal/mol (2.31 Å) | +1.67 kcal/mol (2.08 Å) |
| 9 | True | 4.23 Å | +2.96 kcal/mol (2.37 Å) | +1.51 kcal/mol (2.14 Å) |
| 12 | True | 3.80 Å | +1.95 kcal/mol (2.45 Å) | +1.48 kcal/mol (2.30 Å) |
| 13 | True | 3.67 Å | +2.98 kcal/mol (2.37 Å) | +0.94 kcal/mol (2.10 Å) |
| 14 | True | 3.80 Å | +9.88 kcal/mol (2.62 Å) | +7.60 kcal/mol (2.44 Å) |
Table 1. Comparing ligand models with the bioactive state by energy and geometry.
Use Potentials for Improving Performance on Ligand
The co-folding for Elironrasib in a binary complex with only CYPA was also tested, but "hallucination" became an even more serious issue. None of the models generated the correct ligand stereochemistry. To solve this, I have to add the ‘--use_potentials’ argument, a function in the latest Boltz-2x version designed to steer the diffusion process toward more physically plausible states. This function somehow reduced the hallucination in terms of ligand stereochemistry: now 25 correct models were generated from 175 samples. Interestingly, all 25 reasonable poses were generated from the CCD that was embedded with the bioactive conformation. In other words, the model failed to produce correct stereochemistry from those CCD based on "blind" conformations, which would be the case in a real prediction scenario.
Even for those poses that passed the stereochemistry check, their bi-aryl torsional distribution was still very different from what is seen in the bioactive state of the ternary product (Figure 11a left). Among the 25 generated conformations, only one model had a dihedral angle close to the bioactive state (Figure 11a right). One could argue that a ligand's binary conformation might differ from its ternary conformation (think about tricky linkerology in hetero-bifunctional molecules), but energy calculations suggest that none of the Elironrasib poses generated with CYPA is more stable than the bioactive reference (Figure 11b left). Given the poor diversity of diffused ligand geometries (most pairs have an aligned RMSD < 1.5 Å; Figure 11b right), I think the mechanism hypothesis on Elironrasib as a molecular glue is better to be validated with physical simulations and biophysical experiments.
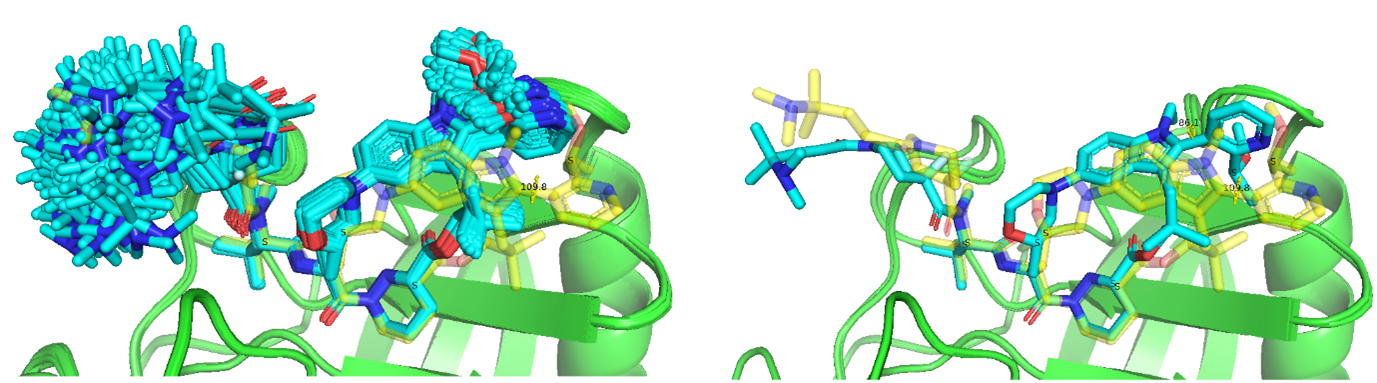
Figure 11a. The co-folding models for Elironrasib (blue stick) with CYPA (green cartoon) under enhanced sampling (–recycling_steps 10 –diffusion_samples 25 for each of 7 customised CCD). Only 1 stereo-correct model has the bi-aryl torsion approaching bioactive state (yellow stick).

Figure 11b. The heatmap of energies and geometries for Elironrasib ligand co-folded within CYPA binary complex. (The bioactive state also include as index 0.)
Then I added the ‘--use_potentials’ function for the generation of ternary complex this time. To my surprise, up to 32 of the 35 Elironrasib samples now have all correct stereochemistry (Recall that only 7 of 15 generated conformations were reasonable without using such physical constraint.) and those correct poses came from various ligand CCD sources. This indicated that the co-folding becomes less sensitive to the input ligand in the presence of more endogenous receptors, which also supported the previous hypothesis that pre-trained models, likely due to their Pairformer architectures, generally prioritise features related to intermolecular recognition over intrinsic physical rules.
Although all generated Elironrasib models had different bi-aryl torsions and showed inferior protein complementarity (in terms of buried solvent-accessible surface area or SASA) compared to the co-crystal structure (Figure 12), some ligand conformations were calculated to be more stable than the bioactive state, based on MMFF-xTB energy minimisations. Given more time, I would also use physics-based methods such as Rosetta, Amber, MMGBSA/PBSA, or FEP to calculate protein-ligand complex and interaction energies. This would allow for a more thorough evaluation of the co-folding model's performance and provide a way to rescue/refine AI-generated structures under physiological conditions (e.g., solvation with water).
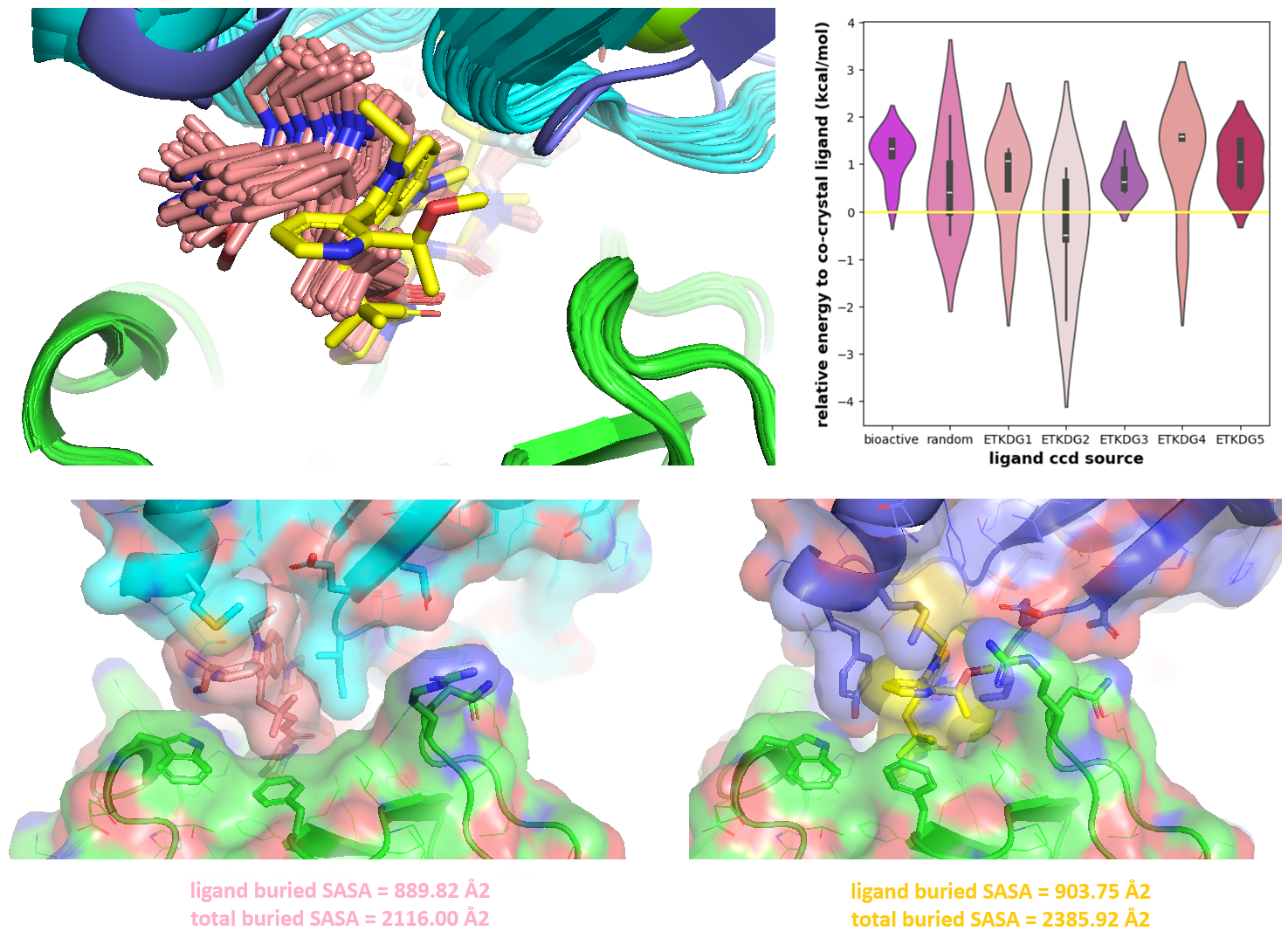
Figure 12. The co-folding performance on ternary complex with ‘-use_potentials’ function in the latest Boltz-2x version. All generated ligand models (pink stick) are analogous and different from the Elironrasib structure (yellow stick) crystallised with CYPA (green cartoon) and KRAS (blue cartoon) in 9BFX. The solvent accessible surface area (SASA) was calculated by PyMOL with settings: dot_solvent 1, solvent_radius 1.4 and dot_density 4.
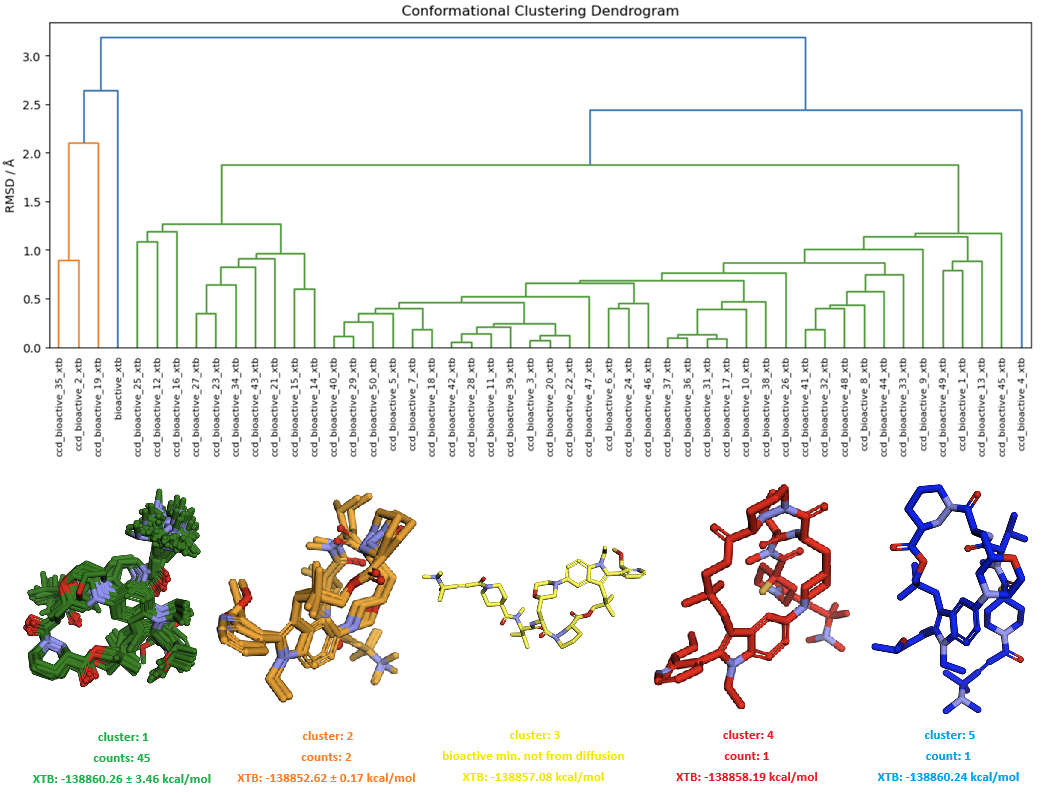
Figure 13. The conformational space sampling for free Elironrasib by diffusion model in Boltz-2x using potentials. All geometries were minimised by MMFF and XTB sequentially, then analysed with hierarchical clustering based on average linkage (RMSD cutoff 2.0 Å).
Outlook
Data and Code Availability
Acknowledgement
This blog post was made possible with excellent open-source projects below:
Boltz: The biomolecular co-folding predictions in this post were generated using the Boltz project. This open-source tool, developed by researchers at MIT, is a cutting-edge deep learning model for biomolecular interaction prediction. I am grateful to the Boltz team for their work.
Website: https://boltz.bioRDKit: The testing heavily utilised the RDKit cheminformatics toolkit for molecular manipulation and analysis. I am grateful to the authors, including Greg Landrum, and the entire RDKit community for their contributions.
Website: https://www.rdkit.orgxTB: All semi-empirical quantum chemical calculations were performed using the xTB program. I extend my appreciation to the developers, particularly the Grimme group, for creating this efficient and accurate method.
Website: https://xtb-docs.readthedocs.ioPyMOL: This work utilised PyMOL for the visualisation of molecular structures. I thank all open-source developers as well as support and maintenance by Schrödinger, Inc.
Website: https://pymol.org